Why I Changed My Mind About Preventing Heart Disease

I have changed my mind about a lot of things over the past two decades of practice. No change has been bigger than how I feel about preventing heart disease.
The medical jargon here is primary prevention. (Re: preventing a first cardiac event). I will tell this story in three chapters.
Chapter 1: What I used to think about primary prevention
In years past, I interpreted the studies of primary prevention as only slightly positive.
Let’s use statins as an example since these drugs are the most studied in all of medicine. Numerous trials have shown that statins consistently reduce the risk of first cardiac events by about 20-25% in relative terms.
Now consider a 45-year-old cyclist with high cholesterol. Should he or she take the drug? In this case, the baseline risk over 10 years of a cardiac event will be low. That means the absolute risk reduction will be tiny: a 25% reduction of a baseline 2% risk (0.25 x .02) is only .5%.
Taking a daily pill in this person reduces his/her risk from 2% to 1.5% over a decade. The number needed to treat or NNT to prevent one cardiac event is 200. That seems quite small.
This framing led me to think we should reserve cholesterol reduction to higher risk people. Because a 25% reduction of someone with a 20% risk over 10 years leads to an absolute risk reduction of 5% not 0.5%. (10x more).
I now consider this wrong thinking.
Chapter 2: What I Believe Now
I now believe that younger lower-risk patients may have the most to gain from primary prevention. An age paradox if you will. Here are three arguments.
First, every statin trial looks like the picture below.
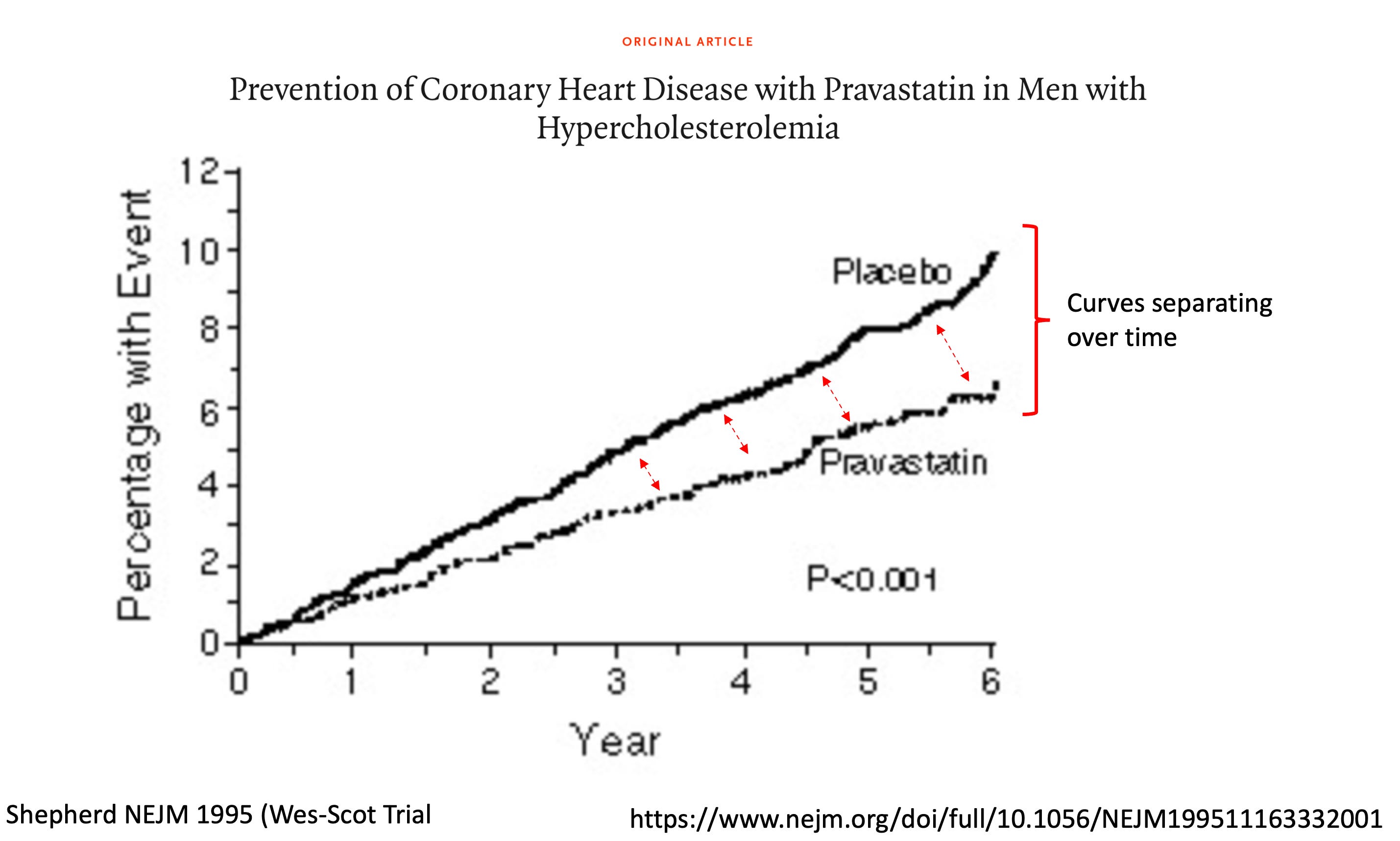
The beneficial effect of the drugs increase over time. At the end of the trial, the effect on reducing events is getting bigger. Cardiology trials usually last 2-5 years. Primary prevention goes a lot longer than that.
Improving effect size over time aligns with our knowledge of atherosclerosis being a gradual disease due to plaque buildup—over years. Diabetes, high blood pressure, smoking, and higher levels of cholesterol all accelerate this process. To wit: the longer that a person is exposed to lower levels of these plaque-promoters, the greater the benefit. This is why teen obesity and diabetes is a pending disaster.
Second, a recent imaging study supports the cumulative idea. (There are many such studies.) Investigators did blood vessel imaging over 6 years in nearly 3500 volunteers. They correlated progression or regression of atherosclerosis with cholesterol and blood pressure levels. Obviously, adults with progression of disease had the highest cholesterol and BP levels.
The most interesting observation was the interaction with age and LDL-C for disease progression. Look at this picture.

It was young people who had the highest odds of progression from elevated cholesterol and BP.
The third reason I believe earlier treatment is better are genetic studies.

These authors took patients who had known gene variants that associate with very low levels of LDL-cholesterol and measured future cardiac events. We call these types of studies Mendelian randomization studies—or, “nature’s randomized trials.”
They allow a comparison between individuals who were “naturally randomized” to having gene variants that cause low cholesterol to people without those variants. Boom. Those with long-term (from birth) exposure to low cholesterol have many fold lower risks of heart disease events.
This graph shows that the effect size of good genes is much larger than those seen in statin trials (wherein cholesterol lowering is started later in life).
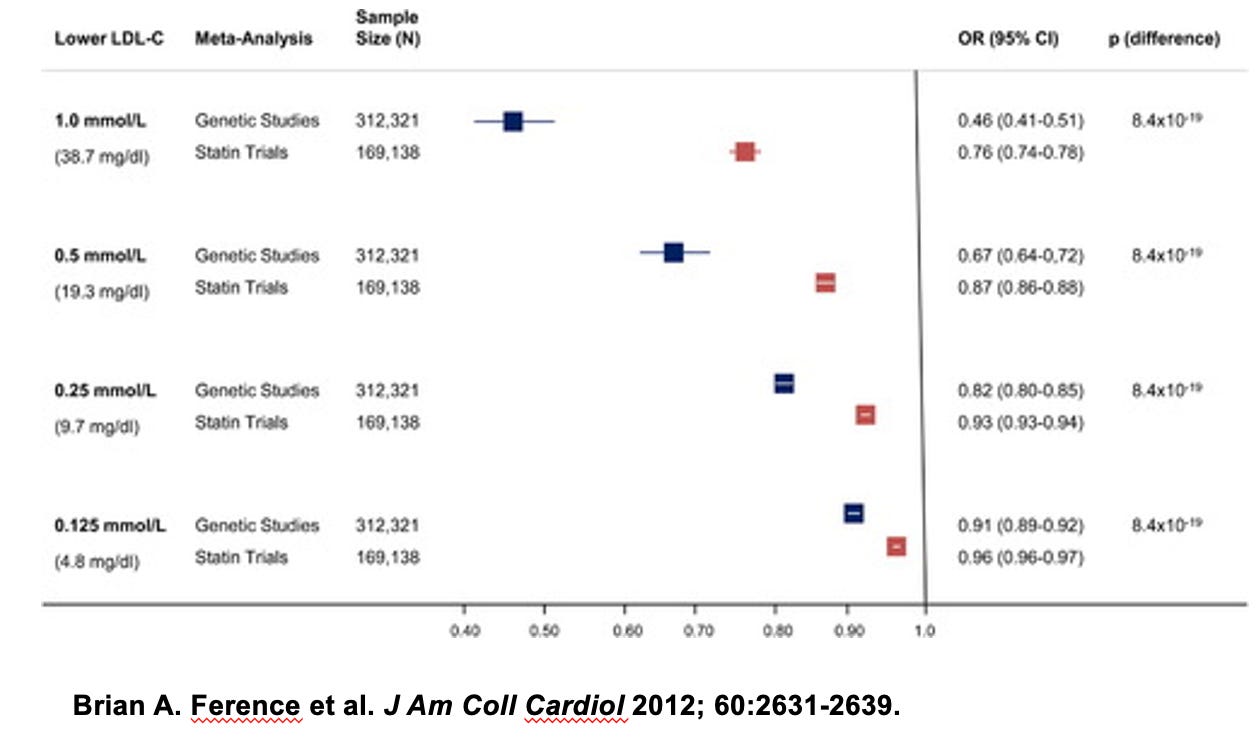
The take home of gene studies are that the longer one is exposed to lower cholesterol the lower the risk of future events. Re: A 45-year-old who takes a statin stands to gain more than a 75-year-old.
The Glue That Holds These Three Ideas Together
This study from Imperial College London researchers glues these three arguments together.

Do read this three-part study. (It’s open access.)
Step one was to use UK mortality data to estimate lifespan gains from a primary prevention therapy (any therapy) that reduced mortality by 30%. (About the same as a statin drug, but this study was not only about statins.)
They then developed a method to calculate the probability distribution of lifespan gain. The math is complex; the idea not so much.
The chief finding was that gains in lifespan from primary prevention are not averaged out over everyone; rather it is concentrated in an unpredictable minority.
This key figure depicts the probabilities of gains from the intervention in a man aged 50 years with average risk. The average lifespan gain is 7 months. But. But. There is a lottery effect. The majority depicted in gray get no gain but the remaining 7% gain a mean of 99 months.

This makes sense, right? If your statin (or BP) pill prevents a major heart attack at age 55, you don’t gain an average of 7 months, you gain many years of lifespan.
The authors also calculated the effect of a primary prevention therapy depending on the age it is started. This gets interesting.
The effect of starting earlier is profound. For example, in men, initiating therapy at age 50, mean lifespan gain available is 12 months with 11% gaining an average of 107 months. If this same group of identical-risk individuals started therapy at 80 years instead, the mean lifespan gain is reduced to 6 months, but those who benefit gain a far smaller amount (56 months).
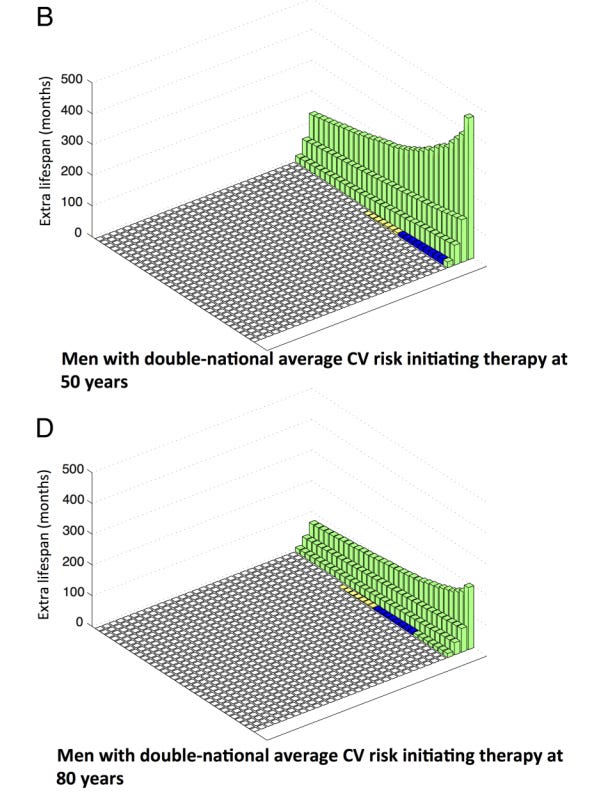
This is the age paradox of primary prevention. I now believe and counsel patients that primary prevention is more likely to be of large benefit when started earlier.
But now to the most important chapter.
Chapter Three — Patients Get to Decide What They Want
The authors of the Imperial College paper I just showed you did a super-interesting thing to assess patient preferences. They went out into public transportation in 3 big UK cities and tested preferences between a certainty of a small gain in healthy lifespan (1 year) versus a percentage chance of a larger gain in healthy lifespan (10 years). (Kahneman and Tversky made these sorts of experiments famous.)
They found that people differed in their preferences. As the percent chance of big gains increased, more people chose that option (white bars). But, still, many people preferred the option that had lower gains. Look at the last column. Nearly one in five people chose a certainty of one year gain (grey bar) even though it calculated to 4 years less survival.

Conclusion:
When I counsel patients about prevention, I explain my change in thinking. There is surely more to gain when we start at a young age.
Don’t misunderstand this as a statin article. I used statins as an example of primary prevention because we know the drugs provide a consistent 25% reduction in future events. You could also insert BP-lowering drugs or cessation of smoking.
Primary prevention can also come in the form of diet and exercise—though we have less empirical data. The advantage of lifestyle intervention is that the unknown unknowns from a taking a drug for decades are absent. And, of course, primary prevention is also not one thing. (To maximize cardiac benefit, patients can do all these things.)
The core principle is this:
Patients have different preferences. Some are minimizers. They will here this message and say no thanks. It’s not worth taking a pill every day. They may look at the many thousand forms of cancer that could get them and say, nope. I am good. Or they don’t like lotteries. That’s ok. It is their choice.
Some patients are maximizers (say, their parents died of heart disease). This group may be happy to hear that earlier therapy is better. And more willing to accept interventions, such as tablets. That, too, is ok.
I see the job of a clinician as that of a paid advisor. I try to explain in a language a patient can understand and then I accept their choice.
JMM
John, I agree with you that primary prevention needs to start earlier in life, BUT the focus should be on the more significant drivers of ASCVD, not cholesterol. The cholesterol-heart theory is a logical fallacy that has been perpetuated for decades.
If cholesterol causes heart disease, then fasting and avoiding sugar and junk food should cause heart disease because that often raises cholesterol, particularly LDL.
Statin trials inadvertently miss a major confounder, insulin resistance, due to the reliance on a glucose-centric metric. LDL....as exciting as it is, with the ability to drive it into the toilet, lacks terrain thinking.
It represents a reductionist lens with which we approach the concept of atherosclerosis causation and prevents the expansion to a more holistic approach; whereby a simultaneous interplay of hormones and the normal state of play of the human body is considered.
You see, insulin resistance is a huge driver generally of high ApoB particle numbers – so the much-lauded correlation between ApoB and poor outcomes…could be mainly due to this aspect. Tricky that. Even after decades of research, I don’t think there’s any data available that tracks ApoB outcomes – specifically when all the other metrics are great, where there is absence of disordered lipid metabolism (either genetically or acquired).
Additionally, the atherogenicity of apoB is not uniform. LP(a) IS AROUND 6-FOLD GREATER THAN THAT OF LDL ON A PER PARTICLE BASIS. Remnants are 5-10 times more atherogenic than LDL. Guess what condition raises remnants? Insulin resistance!
Here are some studies that are relevant:
Effect sizes of low HDL-C and high HDL-C in conjunction with varying levels of TG and LDL-C in the Framingham Offspring cohort showed that LDL levels were irrelevant when TG<100 and high HDL (metabolically healthy).
In the WDHR, 38.7 mg/dL higher LDL-C level was not significantly associated with ASCVD risk in those with CAC=0, whereas it was associated with an 18% increased risk in those with CAC>0. To further support that the presence of coronary atherosclerosis modifies the LDL-C–associated risk for events, a very high LDL-C level (≥193 mg/dL versus <116 mg/dL) was associated with a substantial increased risk of MI and ASCVD in those with CAC>0, whereas no association was found for those with CAC=0. This is in line with previous results from the Multi-Ethnic Study of Atherosclerosis, in which LDL-C level was not associated with future events among asymptomatic individuals with CAC=0 followed for up to 16 years. Indeed, given that atherosclerosis is a multifactorial disease with numerous known risk factors as well as genetic determinants not all patients will develop atherosclerosis despite elevated LDL-C levels. Heterogeneity in the association of LDL-C with coronary artery disease is an inconvenient problem for the "LDL causes ASCVD" crowd. This heterogeneity has also been found in the UK Biobank across the polygenetic background with no association between LDL-C level and coronary heart disease in those with low polygenic cardiovascular risk.
In CLEAR Outcomes study, "Inflammation assessed by hsCRP predicted risk for future cardiovascular events and death more strongly than hyperlipidemia assessed by LDLC." Also, there was no evidence of an association between LDL and cardiovascular death or all-cause mortality.
I have some comments about the studies you reference. There are some irregularities worth noting in the imaging study. There was a high prevalence of central obesity, dyslipidemia (atherogenic), which as stated above, would be consistent with insulin resistance and drive increases in LDL, yet the HOMA-IR was normal. Second, the difference between LDL in the progressors and regressors, was clinically insignificant (136 vs 129).
MR studies rely on strong (often unrealistic) assumptions. One of the many problems with MR is pleiotropy. PCSK9 for example is expressed in many cells and tissues with roles far beyond controlling LDL levels.
Even the supporters of MR have expressed the need for caution. For example, in his recent Editorial, Robert Hegele stated: "Is the assumption of no underlying pleiotropy for any MR that uses common variants ever valid? . . . (1) no scientific method is infallible; and (2) circumspection and vigilance are needed when interpreting MR data." Biology is indeed complex. But this complexity is exactly why we need a better understanding of the disease and its mechanisms, and better controlled studies (with fewer assumptions). These goals are not achieved by MR.
Lastly, a few words about FH. No LDL-C difference exists between FH individuals with and without CVD. No cholesterol-lowering trial has lowered the risk of CVD of people with FH. There are other possible causes. For example. it has been demonstrated that among FH individuals, several factors are
more closely associated with the risk of CVD than LDL-C, and they may indeed be causal.
The commonest and most completely documented are inborn or acquired errors of the
coagulation system and/or other thrombogenic factors, such as increased platelet reactivity.
I am a Hospitalist and come from a health promotion/ disease prevention background. My blind devotion to this mindset has changed considerably with the recognition that net harm may well come as we look to intercede on a younger population, branding the well with pre-illness. John makes a cogent argument here but I would draw these conclusions with great caution. The absolute versus relative risk reduction paradox lives on.